Basic Method Validation
The Detection Limit Experiment
James O. Westgard, PhD
When does a test become less useful? Dr. Westgard gets some help from Karen Mugan, Elsa Quam, Trish Barry, and Neill Carey to explain this confusing aspect of Method Validation. (Preview)
|
|
| Note: This lesson is drawn from the first edition of the Basic Method Validation book. This reference manual is now in its fourth edition. The updated version of this material is also available in an online training program |

MV - The Detection Limit Experiment
Purpose
The detection limit experiment is intended to estimate the lowest concentration of an analyte that can be measured. This low concentration limit is obviously of interest in forensic drug testing, where the presence or absence of the drug may be the critical information desired from the test. Analytical performance at low concentrations is also important for tumor markers, such as prostate specific antigen (PSA), when patient values after treatment may be useful for monitoring "biochemical relapse" [1].
US laboratory regulations require that detection limit (or analytical sensitivity) be verified only for high complexity methods, modified moderate complexity methods, and moderate complexity methods that have not been cleared by FDA as meeting the CLIA requirements for quality control. Given that FDA has not implemented a QC clearance process, the requirement to verify detection limit for moderate complexity methods has been postponed. However until such time that QC clearance has been implemented, good laboratory practice should dictate that detection limit be verified, when relevant, e.g., all forensic and therapeutic drug tests; TSH and similar immunoassay tests; PSA and other cancer markers - and not glucose, cholesterol, enzymes, and constituents where reference range is more relevant for interpretation of the test results.
Terminology in this area is a mess! In making their claims, manufacturers often use a wide variety of terms, such as sensitivity, analytical sensitivity, minimum detection limit, functional sensitivity, limit of detection, and limit of quantitation. At this time there are no accepted standard definitions of these terms, therefore, it is necessary to find out what the actual experimental procedure was, how the data were calculated, how the estimate was made from the data, and whether this estimate is useful for medical application of the test.
Factors to consider
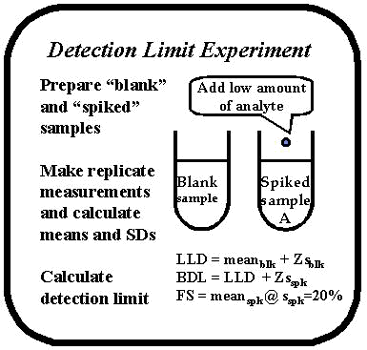 A general description of the experimental procedure is provided in the accompanying figure. Two different kinds of samples are generally prepared. One sample is a "blank" that has a zero concentration of the analyte of interest. The second is a "spiked" sample that has a low concentration of the analyte of interest. In some situations, several spiked samples may be prepared. Both the blank and spiked samples are measured repeatedly in a replication type of experiment, then the means and SDs are usually calculated from the values observed for the samples. Different estimates of detection limit may be calculated from the data on blank and spiked samples.
A general description of the experimental procedure is provided in the accompanying figure. Two different kinds of samples are generally prepared. One sample is a "blank" that has a zero concentration of the analyte of interest. The second is a "spiked" sample that has a low concentration of the analyte of interest. In some situations, several spiked samples may be prepared. Both the blank and spiked samples are measured repeatedly in a replication type of experiment, then the means and SDs are usually calculated from the values observed for the samples. Different estimates of detection limit may be calculated from the data on blank and spiked samples.
Blank solution. One aliquot of the blank solution is typically used for the blank and another aliquot is used to prepare the spiked sample. Ideally the blank solution should have the same matrix as the regular patient samples. However, it is common to use the "zero standard" from a series of calibrators as the blank and the lowest standard as the "spiked" sample.
Spiked sample. In validating the performance of a method, the amount of analyte added to the blank solution should represent the detection concentration claimed by the manufacturer. In establishing a detection limit, it will often be necessary to prepare several spiked samples whose concentrations are in the analytical range of the expected detection limit. For certain tests, there may also be an interest in using samples from patients who are free of disease following treatment (i.e., PSA sera from patients treated for prostate cancer) [2].
We invite you to read the rest of this article.
This article, plus many more important, updated, and expanded chapters are available in the Basic Method Validation manual, 4th Edition

About this website
For over 25 years, WESTGARD QC has provided the latest news, education, and tools in the quality control field. Our goal is to bring tools, technology and training into today's healthcare industry — by featuring QC lessons, QC case studies and frequent essays from leaders in the quality control area. This is also a reference source for quality requirements, including CLIA requirements for analytical quality. This website features the best explanation of the Multirule ("Westgard Rules") and how to use them. For laboratory and healthcare professionals looking for educational and reference material in the quality control field.
THIS IS THE WEBSITE FOR YOU!