Guest Essay
Stability Testing and CLSI EP25-A
A new CLSI guideline has come out with recommendations on the best way to calculate and interpret the stability of IVD reagents. We are pleased to present an essay by James Pierson-Perry, the chairholder of the CLSI subcommittee that developed the guideline, that introduces the concepts and the new recommendations.
Stability Testing of IVD Reagents: Introducing CLSI EP25-A
James Pierson-Perry
Senior Key Expert-Biochemistry
Global Assay Development
Siemens Healthcare Diagnostics
- The "Law of Pharma"
- Elements of Product Stability Testing
- Types of Stability Studies
- Stability Testing Plan
- Measurand Drift Example
- What about Accelerated Stability Testing?
- References
Bias, imprecision, and stability are the fundamental components of most analytical performance attributes for in vitro diagnostic (IVD) reagents. Of these, bias and imprecision are well understood by manufacturers and laboratory scientists. There are internationally accepted protocols for establishment of their claims1,2 and performance verification is done routinely in clinical laboratories.
Stability, in contrast, has greater variability in meaning among both manufacturers and users. This is in large part because stability is not a directly measured characteristic but rather is understood as the capability for a product to retain its stated composition, properties, and performance. As such, stability claims for a product depend on which specific product attributes were assessed over time by the manufacturer. Stability claims are routinely accepted by users from a product’s instructions for use and are only indirectly verified in the laboratory through long-term quality control behavior or similar data.
For this discussion, IVD reagents is taken to include end-use products for diagnostic testing such as reagent kits, calibrators, control materials, sample diluents, etc. The basic stability claims for such products are shelf life—the period of time over which a product remains viable for use when kept under manufacturer recommended storage conditions—and in-use life—the period of time that a product remains viable once placed into use (e.g., opened vial of control material, calibration interval of a reagent kit, etc).
The “Law of Pharma”
There is a wealth of literature reports and regulatory guidance documents relative to assessment of bias and imprecision for IVD reagents. Similar information on stability assessment, however, is considerably more scant. In the US, the FDA requires that IVD reagent manufacturers include stability information as part of their product labeling3 but provides no direct guidance on how to establish such claims. Manufacturers instead are directed to regulations governing drugs and drug substances for information on establishing stability claims4.
In contrast to IVD reagents, the pharmaceutical industry has a great deal of literature on stability experimental design, data analysis, and practical considerations for conducting studies. Key learnings from this knowledge are collected in a series of internationally recognized guidance documents: International Conference on Harmonization of Technical Requirements for Registration of Pharmaceuticals for Human Use.5,6
In 2002, a consensus standard addressing stability testing needs for IVD reagents was released by the European Committee for Standardization. This standard, EN 136407, incorporated best practices from the pharma guidance documents to provide high-level information on terminology, types of stability claims necessary for IVD reagents, and requirements for establishing such claims.
More recently, CLSI released EP25-A: Evaluation of Stability of In Vitro Diagnostic Reagents.8 This document provides a practical implementation of the concepts in the EN 13640 and ICH standards for manufacturers to use in establishment of stability claims for IVD reagents.
Elements of Product Stability Testing
While it is possible to establish stability claims by testing all product attributes over time, a more practical approach is to test only those which are identified as key attributes. This leads to a product specific definition of stability based on three elements:
- selection of the “key” product attributes to be tested
- specification of the associated acceptance criteria
- statement of the desired statistical confidence and power for the analyses
The period of time over which all key attributes meet their acceptance criteria within the desired statistical confidence is taken as the product stability duration. As the components of this definition will vary widely across different products, no single testing protocol would be suitable.
Key attributes should be selected which are most likely to reveal potentially significant changes in product quality, safety, and efficacy during storage and while in use. Manufacturers may choose to draw from physical, chemical, biological, microbiological properties, as well as analytical performance indicators.
One of the most common key attributes is the apparent change in measured quantity of an analyte over time, referred to as measurand drift. This may be used to assess either actual changes in concentration (e.g., for control products or calibrators) or changes in measurement bias of a method (e.g., calibration interval) over time. Such changes may be measured on an absolute basis or as relative basis referenced to product maintained under presumed stable conditions.
Suitable acceptance criteria for key attributes may come from multiple sources: product design input requirements, established quality goals for methods (e.g., CLIA, RiliBÄK), historical data of similar products, etc. Quite often, multiple test samples will be used to assess analytical performance attributes over the method range. In these cases, the minimum stability duration for any one sample should be used as the overall stability duration for that product lot.
CLSI EP25-A requires that establishment of stability estimates be based upon considerations of statistical confidence and power for creating experimental protocols and subsequent data analyses. The number of replicates and timing of test points should be chosen so as to adequately discern trending from variability of the results.
Types of Stability Studies
Both EN 13640 and CLSI EP25-A describe the need for different types of product stability testing. Shelf-life studies are used to establish expiration dating for an IVD reagent in the final packaging under defined storage conditions. This represents the interval from product manufacture to the final day of usability—whether or not it is ever actually put into use. The guidance documents require that three product lots be used to establish shelf-life claims.
In contrast, in-use claims define the period of time in which the product remains suitable after being placed into use. Example of such claims include the duration that a control product may be used after being first opened or that a method may be used prior to needing routine recalibration. A single product lot may be used to establish these claims but it is wise to use more.
The third type of testing is known as transport simulation or, more simply, stress testing. These studies involve an exposure of sample product to sets of predetermined changes in environmental conditions (e.g., temperature, humidity, light exposure, etc.) over time in order to simulate worst case conditions that a product might endure during its distribution cycle prior to customer receipt. Results of such studies help identify appropriate shipping and handling conditions for a product. The stress testing conditions used may come from a manufacturer’s own studies on actual product distribution or from references such as ASTM:D4169-05.9 At least one product lot must go through this testing.
Stability Testing Plan
CLSI EP25-A requires establishment of a stability testing plan prior to the start of any testing. This plan documents what is to be tested, experimental protocols and details for the testing, and acceptance criteria. Typical expected elements of the plan include:
- Identification of the product
- Which type(s) of stability studies will be done
- Product attributes to be tested
- Acceptance criteria for each test
- Number of product lots to be tested
- Materials and experimental designs to use for testing
- Sampling plan (i.e., how to select representative product samples from a lot)
- Testing schedule
The testing schedule should extend beyond the desired stability duration. This will ensure that the estimate falls within the testing period with no extrapolation and takes into account that consideration of confidence intervals often leads to more conservative stability estimates than would be obtained otherwise. It is expected that sufficient testing replicates will be used to ensure adequate statistical power of the stability estimates.
There are two basic approaches to design of stability studies. The classical approach has product put into the desired storage conditions on Day 0, then testing is done per schedule with product taken from storage. There are no prerequisites for this design and interim results are available following each testing point. A disadvantage of this design is its susceptibility to influence from variability in measurement processes over the study duration and bias from changes in measurement processes due to instrument maintenance activities, reagent lot changes, or component failure.
An alternate approach is the isochronous design.10 Here, product also is put into storage on Day 0 but on each day of the schedule, product is simply removed from storage and put into presumed stable storage conditions (e.g., -70 ºC). At the end of the study, all product samples are taken and run in a randomized order as a single batch. This design greatly minimizes variability and bias of the measurement process, relative to the classical design. Disadvantages are that stability estimates are not available until the final testing point and that a stable interim storage condition must be available. This experimental design is particularly useful with studies of short duration (e.g., control product in-use stability).
Measurand Drift Example
The following example, taken from CLSI EP25-A8, illustrates the impact that consideration of statistical confidence may have on establishing shelf-life stability by measurand drift.
A new IVD reagent for ferritin was used to test several quality control and human serum pool samples, with concentrations that fell across the assay range, over a duration of 402 days. At the end of the study, the mean observed [ferritin] values versus test days were analyzed by standard linear regression for each sample. The p values for all slopes but one were > 0.15, indicative of no significant change over the testing period. The one sample with a significant slope (p < 0.001) was for a serum pool near the high end of the assay range.
The measurand drift acceptance criterion was ± 5%. As the Day 0 value for this sample was 1855.0 µg/L, the allowable upper limit for observed ferritin concentration was 1947.8 µg/L. The data were plotted and an upper 95% confidence interval was constructed about the regression line. A one-sided confidence limit was used as the expected positive increase in observed results was anticipated from previous studies. A stability estimate of 395 days was identified at the intersection of this confidence interval with the maximum allowable ferritin concentration.
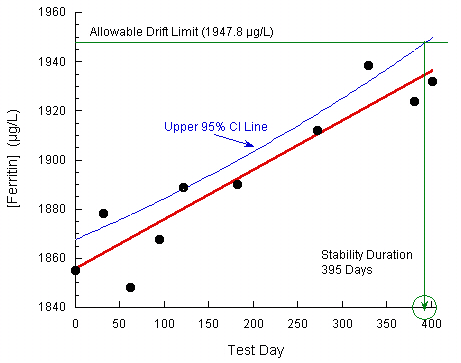
Plot taken from CLSI. Evaluation of Stability of In Vitro Diagnostic Reagents; Approved Guideline. CLSI document EP25-A. Wayne, PA: Clinical and Laboratory Standards Institute; 2009. Reprinted with permission.
If the confidence interval is ignored, the stability estimate is computed from the regression fit statistics (slope = 0.2011, Y-intercept = 1855.0) as:
(Allowable Drift Limit – Y-intercept)/Slope = Stability Estimate
(1947.8 – 1856.0)/0.2011 = 461 days
As expected, use of the confidence interval results in a significantly more conservative stability estimate than otherwise.
What About Accelerated Stability Testing?
Accelerated stability testing refers to the use of more stressful storage conditions than normal in order to cause product degradation on a faster time scale than would normally occur. Such studies may be useful for:
- comparing the relative effectiveness of variant product formulations or packaging options
- identifying modes of instability for product risk analyses
- assessing the impact of proposed changes to product design or manufacture
- providing early estimates of product stability
A significant concern with accelerated testing is that application of stress storage conditions could cause product instability through mechanisms that would not occur under normal storage conditions. In addition, not all products will follow a predictable failure trend and might show acceptable performance up to the moment of failure.
While accelerated testing can be of great use when making product design decisions, the general expectation of regulatory agencies is that product stability claims will be based on real-time data. There are cases where accelerated testing data alone has been sufficient but, more often than not, such data are expected to be followed with final results from real-time studies. It is good practice to check first with relevant regulatory agencies on what stability data would be required for a specific product.
References
- CLSI/NCCLS. Evaluation of Precision Performance of Quantitative Measurement Methods; Approved Guideline—Second Edition. CLSI/NCCLS document EP05-A2. Wayne, PA: NCCLS; 2004.
- CLSI/NCCLS. Method Comparison and Bias Estimation Using Patient Samples; Approved Guideline—Second Edition. CLSI/NCCLS document EP09-A2. Wayne, PA: NCCLS; 2002.
- Code of Federal Regulations, 21 CFR Part 809.10 (a) (5). Labeling for in vitro diagnostic products.
- Code of Federal Regulations, 21 CFR Part 211.166. Stability testing
- Expert Working Group (Quality) of the International Conference on Harmonization of Technical Requirements for Registration of Pharmaceuticals for Human Use. Guidance for Industry. ICH Q1A (R2) Stability Testing of New Drug Substances and Products. Step 4. February 2003.
- Expert Working Group (Quality) of the International Conference on Harmonization of Technical Requirements for Registration of Pharmaceuticals for Human Use. Guidance for Industry. ICH Q1E Evaluation of Stability Data. Step 4. February 2003.
- CEN. Stability testing of in vitro diagnostic reagents. EN 13640. Brussels: European Committee for Standardization; 2002.
- CLSI. Evaluation of Stability of In Vitro Diagnostic Reagents; Approved Guideline. CLSI document EP25-A. Wayne, PA: Clinical and Laboratory Standards Institute; 2009.
- ASTM. Standard practice for performance testing of shipping containers and systems. ASTM D4169-05. West Conshohocken, PA: American Society for Testing and Materials; 2007.
- Lamberty A, Schimmel H, Pauwels J. The study of the stability of reference materials by isochronous measurements. Fresenius J Anal Chem. 1998;360:359-361.